定点突变
实验目的
我要克隆一个老鼠里的基因的CDS出来,与NCBI上下载的参考序列相比,发现在其第12个氨基酸处存在一个单碱基突变,引起了编码的氨基酸改变。有没有必要修过来呢?不同的人意见不同,有人说:
“不同的研究者需要有个共同的标准,即便你能保证是从老鼠里原模原样克隆出来的,也最好跟数据库统一一下。”
也有人说:
“完全没有必要呀,数据库中的序列只有一套,而一个基因显然在不同的种群和个体中有不同的等位基因,这种单碱基突变太正常了,不用修。或者有空就修,没空就不修。”
这个问题我还没有一个定论,总之我这次是选择修了。
第一次尝试
我的目的是给该基因CDS的N端加上3xFlag,同时修复第12位氨基酸的突变。为了省事,我直接把这两件事设计在了一条引物上,即引物5'端有一段长长的3xFlag,而3'端的最后一个碱基刚好覆盖突变位点。没有再往后延伸是因为这个引物已经80多bp了,我当时认为只要最后有一个碱基覆盖突变位点,就能通过PCR修复过来,毕竟,PCR得到的所有产物都是从引物的3'末端开始延伸的。

结果是,所有拿去测序的几个转化子都没有把这个突变位点修复过来,我郁闷了好几天,还问了祥鸿是不是有引物合成错误的可能。有个技术人员跟我打电话说我做点突变的方式不对,他们做突变引物的时候会在突变位点两端都添加15bp以上。可是,当我问到“PCR产物每一天都是从引物开始延伸的,从理论上不可能修复不过来”时,他也没有好的解释。
有天晚上跟光老师吃饭时谈到了这个问题,第二天他告诉我,有可能是我使用的高保真酶拥有3'→5'的校对活性,因为最后这个碱基配对不上所以被切掉了,我没有做实验验证,但是我觉得他是对的。回头想想,就算没有校对活性,我把一个与模板不配对的碱基放在引物3'端也是很愚蠢的行为,毕竟影响配对了,谁知道会如何影响结果呢。
第二次尝试
虽然点突变没修过来,但是我的3xFlag-tag已经加好了,所以我想在这个质粒的基础上,通过环状PCR的方式来修复这个突变。

第二次尝试的结果是:我一共前后送测了8个质粒(N端加Flag和C端加Flag一共),其中5个突变修复了,3个突变未修复。但是,5个修复了的转化子在引物两侧100bp左右全部存在约100bp量级片段的插入,而剩余3个突变未修复的转化子中,也有1个存在这样的插入片段,剩下两个片段是对的,就是没有修过来突变。
从上图可以看到,我用的方法是设计一对完全反向互补的引物,突变位点在正向引物的上游,反向引物从3'端开始数的第七个碱基。PCR之后用DpnI消化甲基化模板,直接转化涂布。这次突变修复成功的频率是5/8,可见之间确实有可能是因为3'端不配对被高保真酶切掉。但有6/8的质粒成环后发生了片段插入,我后来根据测序结果中高概率发生插入的地方设计了引物,又挑了一些菌落PCR鉴定,发现大小真的是群魔乱舞。
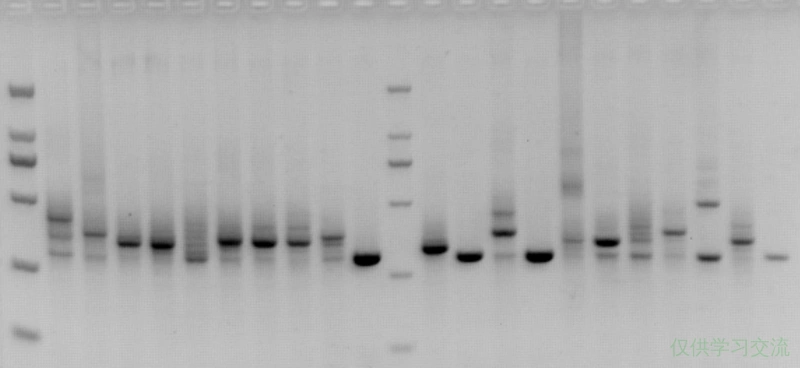
质粒环P是个很经典的方法,不应该成功率这么低,可能还是我设计的引物太短了。
第三次尝试
后来,MRZ师兄建议我设计引物时再长一点,同时不要完全反向互补,上下游引物要交错开。我新的引物在突变位点两翼各延伸了15bp。
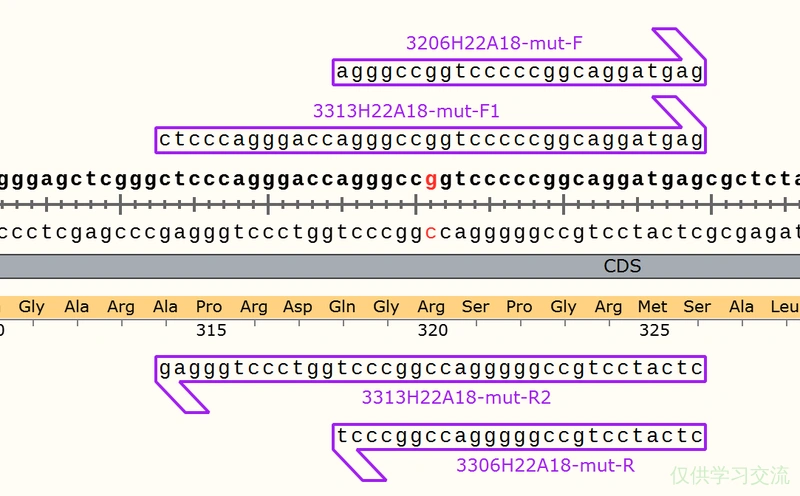
不过因为构建这个克隆已经失败两次,耽误了太多时间,所以我就没有继续尝试环P的方法了,而是用步骤更多但是成功更有把握的方式,在质粒对侧也设计了两条引物,分两个PCR反应得到两端具有同源臂的片段,利用无缝克隆技术连成环了。
第三次尝试的结果是,N端加Flag的质粒送测了两个转化子测序没问题,突变修复过来了,而且质粒成环也没问题,但是C端加Flag的质粒送测了4个菌落,它们的突变位点都没有修复过来,还是原来模板上的碱基,这个就很离谱了。如果是PCR酶的校对活性导致之前修复不了的结果,这次我在突变位点两侧都加长了引物,它难道能连续校对切割16个bp吗?
第四次尝试
所以说,为何突变不过来的问题我依然没有一个准确的解释原因,我也没有再花经历去探究为什么,因为N端加Flag的质粒已经构建好了,所以我直接从这个质粒下PCR下来已经修复突变的完整CDS、用双酶切连接法加上了C端Flag,这个事情就结束了,伴随着为何突变这么难修复的谜题。
融合PCR
实验目的
从人的cDNA中克隆一个蛋白的CDS,换了几次PCR条件一直都没P出来,后来才意识到,我从NCBI上下载的序列可能来自一个表达并不高的isoform (isoform a, 最长的isoform),而模板中表达更高的是isoform b,isoform b因为可变剪接的原因在N端比isoform a少了几十个氨基酸,因此我的引物根本mapping不到cDNA模板里的isoform b上,自然P不出来。这是我第一次严肃意识到实验中对RNA和蛋白剪接变体进行选择的问题。我后来跟几个人分别谈起过这个问题,一部分人的选择都是无脑选择最长的那个(这次我也一样),因为最长的变体至少意味着不会因为氨基酸片段缺失导致功能不完整。但这种想法显然是有风险的,我们无法准确预计不同isoform之间相差的肽段的功能是什么,如果恰好缺少的部分是一个自抑制功能的domain,那这两个isoform的功能就会完全相反了。这个问题也暂搁置,我这次是既克隆了最长的isoform a,也克隆了isoform b。Isoform b从cDNA中换引物克隆即可,但是isoform a从cDNA中得不到,因此我请公司合成了isoform b相比isoform a的N端短的那一部分,再自行把它和isoform b连接起来(考虑到价格问题没有直接全合成isoform a)。所以抽象一下问题就是,有两段DNA序列分别在两个质粒中,我需要把它无缝拼接起来。用的方法是融合PCR,因为第一次做融合PCR,所以记录一下。也挺简单的,临时百度了一下,一次就成功了。
实验方法

- 两个引物对分别是F1,R1和F2,R2,先各自PCR出片段(有30多bp重叠部分),然后胶回收。
- 胶回收产物大致计算摩尔比1:1混在一起,配成25微升不加引物的PCR体系,进行5个cycle的PCR。
- 直接开盖加入另外25微升两倍引物(F1和R2)、不加模板的25微升PCR体系,PCR 35 cycles,胶回收。
- 下游的双酶切、连接、转化……
两个以后要继续讨论的问题
- 怎样看待手中的序列与数据库参考序列不一致的问题。
- 怎样选择不同RNA剪接变体和蛋白变体作为研究对象。
- 定点突变技术还要学习。
有话这里说 ↓